Abstract
In recent years, and even more since its legalization in several jurisdictions, cannabis and the endocannabinoid system have received an increasing amount of interest related to their potential exploitation in clinical settings. Cannabinoids have been suggested and shown to be effective in the treatment of various conditions. In cancer, the endocannabinoid system is altered in numerous types of tumours and can relate to cancer prognosis and disease outcome. Additionally, cannabinoids display anticancer effects in several models by suppressing the proliferation, migration and/or invasion of cancer cells, as well as tumour angiogenesis. However, the therapeutic use of cannabinoids is currently limited to the treatment of symptoms and pain associated with chemotherapy, while their potential use as cytotoxic drugs in chemotherapy still requires validation in patients. Along with cannabinoids, cannabis contains several other compounds that have also been shown to exert anti-tumorigenic actions. The potential anti-cancer effects of cannabinoids, terpenes and flavonoids, present in cannabis, are explored in this literature review.
1. Introduction
Archaeobotanical evidence and written records found in ancient texts of Ayurvedic medicine and the first known Pharmacopoeia “Shen Nung Pen Ts’ao Ching” describe medical use of cannabis for several thousand years. Cannabis use, for religious/spiritual, food, and textile has been documented in written history back to at least the third millennium BC, and potentially even earlier by archaeological evidence (history reviewed in [1,2]). Cannabis reached South America in the mid-1500s and North America in the early 1600s. During the development of Western Medicine, a progressive understanding of cannabis properties led to wider medical and recreational consumption in the 20th century until the use of this plant became marginalized and criminalized, largely due to misinformation, which greatly impacted progress regarding the understanding of the medicinal benefits of this plant and its components [3,4].
The canonical endocannabinoid system is comprised of the main endocannabinoids anandamide and 2-arachidonoylglycerol and cannabinoid receptors CB1 and CB2. Additional components, such as the endocannabinoid-degrading enzymes fatty acid amide hydrolase and monoacylglycerol lipase, other cannabinoid-activated G protein-coupled receptors and members of the transient receptor family, among others, could also contribute to the effects of cannabinoids, and are therefore identified as possible targets involving this class of compounds. The effects of the endocannabinoid system and its potential involvement in cancer have been discussed in several recent publications and only some highlights of the general functions and effects related to cancer are therefore provided here [5,6,7]. Dysregulation of the endocannabinoid system has been implicated in several diseases, including cancer. This dysregulation can include variation in the expression and/or function of cannabinoid receptors and enzymes, or simply alterations the concentration of endocannabinoids [8,9]. For example, dysregulation of cannabinoid receptor levels in malignant tissues has been observed [10] and was associated with poor prognosis for patients with different types of cancer [11,12,13,14,15,16,17]. The levels of endocannabinoids have also been shown to be dysregulated in malignant tissues. For example, concentrations of AEA and 2-AG were increased in colorectal carcinomas when compared with healthy neighboring tissue [18,19,20,21]. Early investigations into the functional implication of endocannabinoids during tumor progression demonstrated that endocannabinoids had inhibitory effects on the proliferation of breast or prostate cancer cells [22,23]. Based on the preliminary evidence in various models, it appears that cannabinoids target key signaling pathways involved in all the hallmarks of cancer [24]. In several indications, cannabinoids complement conventional chemotherapeutic regimens by reducing some of their adverse effects such as pain, nausea, and vomiting. Additionally to the cannabinoids, a large number of terpenes and flavonoids, some of them also present in cannabis, exhibit cytotoxicity against a variety of cancers [25,26]. Fundamental research will allow us to better understand the interrelationship between the various compounds present in the cannabis plant, the endocannabinoid system and cancer. Research will therefore help identify intracellular signaling pathways that participate in cannabinoid anticancer action, and will help discern in which circumstances these compounds should be best tested in clinical trials (reviewed in [27]) for eventual therapeutic use; whether as single therapy agents, synergistically with a validated chemotherapeutic agent or in polypharmaceutical formulations. Below is summarized the current knowledge about the potential effects of cannabinoids, terpenes and flavonoids present in cannabis, as anticancer agents.
2. Cannabinoids
More than 100 cannabinoids have been isolated from the plant Cannabis sativa [28]. Cannabinoids derive from cannabigerolic acid and differ mainly in the way this precursor is cyclized (Figure 1). Phytocannabinoids can be found in other plant species besides cannabis. These include several types of Echinacea, Acmella oleracea, Helichrysum umbraculigerum and Radula marginata [29]. Due to its psychoactive effects, the phytocannabinoid tetrahydrocannabinol (THC) is the best-known phytocannabinoid and the primary intoxicating compound in cannabis. Cannabinol also displays intoxicating effects. Most other phytocannabinoids are not intoxicating, the best known being cannabidiol, but also include others, such as cannabigerol, cannabivarin, cannabichromene. The effects of cannabinoids have been examined for various conditions, and we highlight here some of their effects in cancer (Table 1). Considering all the available literature at this time, much stronger experimental evidence (obtained in vitro, in vivo and even in a few clinical trials) support that THC and cannabidiol (CBD) have better anticancer activity than for the other cannabinoids.
| Compound | In Vitro Effects | In Vivo Effects | Clinical Trials |
|---|---|---|---|
| Δ9-Tetrahydrocannabinol | |||
| Breast Cancer | Inhibited cell growth and proliferation [30,31,32]. Inhibited estradiol-induced proliferation [31,33]. Increased proliferation and tumor growth [34,35]. Activated transcription factor JunD [32]. Induced apoptosis and cell cycle arrest at G2/M phase [36]. Induced fatty acid 2-hydroxylase [37]. Increased production of reactive oxygen species [38]. Inhibited human P-glycoprotein and breast cancer resistance protein [39]. |
Increased tumor growth and metastasis [35]. Reduced tumor growth [37,38,40,41]. Inhibited tumor angiogenesis [40]. |
N/A |
| Brain Cancer | Inhibited cell viability and proliferation dose-dependently [42,43,44,45]. Induced apoptosis [46,47,48,49]. Stimulated glioma cell growth [50]. Induced autophagy via ceramide accumulation and ER stress [47,51,52]. Down-regulated expression of matrix metalloproteinase-2 [53]. THC + CBD pre-exposure increased sensitivity to radiation therapy [53]. |
Reduced tumor growth [47,48,51,52]. Upregulated stress protein p8 and induced apoptosis [47]. Induced autophagy [48]. THC + Temozolomide synergistically reduced growth of xenograft tumors [54,55,56]. Down-regulated expression of metalloproteinase-2 [53]. THC-loaded nanoparticles reduced cell proliferation, angiogenesis and increased apoptosis [57]. |
Pilocytic astrocytoma tumors regressed over a period of 3 years following the inhalation of cannabis over the same period [58]. Temozolomide + Sativex increased 1-year survival rate in GBM patients [NCT01812603 and NCT01812616]. Reduced tumor cell Ki67 staining in patients suffering from recurrent GBM [59]. Reduced VEGF and VEGFR-2 activation in GBM patients [60]. Dronabinol treatment did not lead to severe adverse effects in patients with primary brain tumors [61]. |
| Leukemia | Reduced proliferation and exhibited cytotoxicity [62]. Sensitized leukemia cells to anti-cancer agents [62,63,64]. Inhibited the differentiation blockage (Dronabinol) [65]. Induced apoptosis [66,67,68]. Induced apoptosis in patient-derived leukemia cells [66] |
N/A | Remission achieved following the consumption of Cannabis sativa oil in a patient with terminal acute lymphoblastic leukemia [69]. Dronabinol inhibited the differentiation blockage in leukemia patients [66]. |
| Lung Cancer | Low levels induced cell proliferation or did not decrease cell survival [50,70]. Inhibited cell proliferation, chemotaxis and invasion [71,72]. Reduced migration [72]. Inhibited host immune response and killing of tumor cells [73]. Suppressed EMT of NSCLC cells [72]. THC-loaded nanoparticles exhibited cytotoxicity [74]. |
Increased tumor growth and reduced tumor immunogenicity [75]. Inhibited tumor growth and metastases [71]. THC-loaded nanoparticles exhibited significant cytotoxicity [74]. |
N/A |
| Melanoma & Myeloma | Inhibited growth and proliferation [76,77]. Induced apoptosis and autophagy [78]. Induced autophagic-dependent necrosis [77]. THC + CBD had synergistic effects with carfilzomib [77]. Increased cell death and decreased migration [77]. |
Reduced proliferation, metastasis, angiogenesis, tumor growth and increased apoptosis [76,79]. THC:CBD in a 1:1 ratio decreased tumor growth and increased autophagy and apoptosis [78]. THC + Trametinib reduced viability, invasion and metastasis of MEKi-resistant melanoma cells [80]. Induced myeloid-derived suppressor cell function and differentiation [81]. |
N/A |
| Hepatocellular Carcinoma | Decreased cell viability and induced autophagy [82]. Increased activity of PPARγ [83]. Reduced proliferation, migration, invasion, and induced apoptosis [84]. |
Reduced tumor growth [82]. Increased the activity of PPARγ [83]. THC + Irinotecan reduced hepatic toxicity during acute treatment [85]. |
N/A |
| Pancreatic, Prostate, Colon Cancer | Decreased cell viability [30,86,87]. Induced apoptosis [86,88,89]. THC-loaded microspheres inhibited proliferation [90]. |
Reduced the growth of tumors [86]. | N/A |
| Endometrial, Cervival, Oral Cancer | Increased accumulation of anti-cancer agents in cells expressing multi-drug transporters [91,92]. Reduced invasion via increased TIMP-1 expression [93]. Inhibited mitochondrial oxygen consumption and exhibited strong toxicity [94]. |
N/A | N/A |
| Cannabidiol | |||
| Breast Cancer | Induced apoptosis and autophagy [30,95]. Enhanced production of reactive oxygen species and subsequent ER stress [95]. Inhibited proliferation, migration and invasion [96,97,98]. Inhibited the EMT and reduced expression of malignant markers [98]. Increased sensitivity to anti-cancer agents doxorubicin and cisplatin [98]. Synergistic effects with paclitaxel and doxorubicin on antiproliferative activity [99]. |
Inhibited tumor growth, migration, invasion, and metastasis [97]. Increased survival and decreased metastasis [100]. Down-regulated Id1 expression [100]. |
N/A |
| Lung Cancer | Induced apoptosis [101]. Reduced invasion, metastasis, migration, and restored epithelial phenotype [72,102,103,104]. Increased susceptibility to lysis by lymphokine-activated killer cells [105]. |
Reduced cell viability [101]. Decreased tumor growth [101,103]. Decreased metastasis [103]. |
N/A |
| Glioma & Neuroblastoma | Inhibited cell proliferation and induced apoptosis [43,106,107,108,109,110,111]. Increased reactive oxygen species production [110,112]. Increased expression of heat shock proteins [112]. Induced cell cycle arrest [111]. Reduced invasion [109,111]. |
CBD + THC + Temozolomide reduced tumor growth [54,55]. Reduced tumor growth [57,108]. Enhanced apoptosis and decreased angiogenesis [57]. Significantly prolonged mouse survival [110]. |
Sativex + Temozolomide increased the rate of 1-year survival by 39 percent in GBM patients [NCT01812603 and NCT01812616]. |
| Colon & Prostate Cancer | Induced apoptosis, cell cycle arrest and ROS production [87]. Reduced cell proliferation, promoted apoptosis and elevated ROS levels [87,113,114,115,116]. Antagonistic activity at GPR55 reduced and prevented metastasis [117]. |
Increased effects of anti-cancer agents bicalutamide and docetaxel [87]. Reduced aberrant crypt foci polyps and tumor growth [87,113,114,118]. Chemo-preventative on colon cancer cells due to up-regulated caspase-3 [113]. Decreased metastasis and angiogenesis [114]. |
N/A |
| Myeloma, Melanoma, Leukemia | Reduced cell viability [77,119]. Induced apoptosis due to ceramide accumulation [120]. Decreased P-glycoprotein expression and sensitized cells to Vinblastine [64]. Increased cytotoxicity of bortezomib and carfilzomib [77,119]. |
Increased mouse survival and reduced tumor growth [78,116]. | N/A |
| Cervical, Endometrial, Ovarian Cancer | Inhibited cell growth and induced apoptosis [121,122]. Increased intracellular accumulation of multi-drug transporter substrates Fluo3, vincristine, and mitoxantrone [91,92]. |
N/A | N/A |
| Cannabigerol | Significant inhibitory effects on cell proliferation [123,124,125]. Inhibited [14C]anandamide uptake and activated TRPV1 receptor [30]. Stimulated apoptosis and ROS production [123]. |
Decreased tumor growth due to antagonistic activity at TRPM8 receptors [123]. | N/A |
| Cannabichromene | Inhibited cell viability and growth [30,87,123]. Significantly activated caspase 3/7 [87]. Elevated intracellular Ca2+ levels [87]. |
N/A | N/A |
| Cannabidivarin | Dose-dependent inhibitory effects on cell viability [87,123]. | N/A | N/A |
| Cannabinol | Cytotoxic effects at high concentrations [87]. Antiproliferative effects [96]. Inhibited multi-drug transporter ABCG2 and promoted accumulation of mitoxantrone [92]. |
N/A | N/A |
| Tetrahydrocannabivarin | Cytotoxic effects at higher concentrations [87]. | N/A | N/A |
2.1. Delta9-tetrahydrocannabinol (THC)
Δ9-tetrahydrocannabinol (THC) is the major psychoactive component present in Cannabis sativa L. cultivars, mediating its effects in the central nervous system via CB1 receptors [126]. THC binds and activates CB1 receptors in the central nervous system (CNS), leading to the intoxicating feelings associated with cannabis use. THC can be administered via multiple routes, including orally, intravenously, intramuscularly and inhalation. The most common method of administration in humans is orally, and due to its high lipophilicity, it is highly bound by plasma proteins and is readily distributed to vascularized tissues such as the liver, heart and lungs. Fat tissues have also been shown to be reservoirs for THC accumulation. Due to the psychoactive effects of THC mediated in the CNS, there are concerns in terms of prescribing THC for medicinal use in cancer patients. There are also other undesirable side effects of THC use, such as dependence, tolerance and issues surrounding abuse [27]. Despite the limitations and concerns associated with THC treatment, there are many studies regarding THC’s potential as an anti-cancer therapy and we highlight these studies herein.
2.1.1. Breast Cancer
In breast cancer cells, THC at a concentration of 14 µM inhibited overall cell growth and proliferation [30]. Exposure to THC was shown to inhibit estradiol-induced cell proliferation by inhibiting estrogen receptor α activation [31]. THC exposure antagonized 17β-estradiol-induced proliferation, and did not act on androgen or estrogen receptors in MCF-7 cells [33]. In contrast, Takeda et al. found that THC increased human epidermal growth factor 2 (HER2) expression, which is able to stimulate cancer cell proliferation, and that THC had proliferative actions in MCF-7 cells [34]. Similarly, a study by McKallip et al. [35] found that treatment of tumors with low levels of cannabinoid receptor expression with THC can actually lead to increased tumour growth and did not induce cytotoxicity in these cells. In addition, they showed that 4T1 mouse mammary carcinoma cells were also resistant to THC, and treatment of these cells in vivo with THC resulted in increased tumor growth and metastasis as a result of suppression of the specific anti-tumor response. Mechanistically, THC’s anti-cancer effects in breast cancer can be mediated by modification of JunD, a transcription factor. THC was shown to activate JunD by both translocating it to the nucleus and up-regulating its expression [32]. This was confirmed by testing THC in breast cancer cells with silenced JunD and JunD knockout mice-derived fibroblasts, where the anti-proliferative effects of THC were significantly reduced. Another study showed that THC reduced human breast cancer cell proliferation via stimulation of CB2 receptors. THC treatment inhibited the cell cycle progression in breast cancer cells at the G2/M phase, which was attributed to the down-regulation of Cdc2, and induced apoptosis [36].
The ability of THC to treat ErbB2-positive breast cancer, a very aggressive form of cancer has been evaluated. In a mouse model of ErbB2-driven metastatic breast cancer, THC treatment was able to reduce tumor growth, as well as the amount and severity of lung metastases. THC treatment also induced apoptosis and limited tumor angiogenesis [40]. Heteromerization of HER2 receptors with CB2 receptors has been shown to control the oncogenic activity of HER2 and is connected to poor patient prognosis [41]. THC treatment disrupted HER2-CB2 receptor heteromers via the binding of CB2, which ultimately resulted in anti-tumor actions both in vitro and in vivo. In a xenograft murine model, THC treatment significantly reduced tumor growth and resulted in decreased expression of the HER2 protein. In MDA-MB-231 breast cancer cells, THC was shown to cause fatty acid 2-hydroxylase (FA2H) induction [37]. A possible mechanism mediating this increase in FA2H is the induction of peroxisome proliferator-activated (PPAR) α [127]. Higher levels of FA2H expression has been reported to be a crucial biomarker that is related to the prognosis of patients with triple-negative breast cancer, where higher levels of FA2H has been shown to result in shorter disease-free survival [37]. Recently, the effects of pure THC versus a botanical drug preparation in the treatment of breast cancer were evaluated. It was found that the whole botanical drug preparation was more potent in terms of anti-tumor action in cellular and murine models of breast cancer. In vivo, THC was found to be less potent at inhibiting tumor growth than the botanical extract. Pure THC produced anti-cancer effects via CB2 and the production of reactive oxygen species, and the botanical preparation exerted its effects through different mechanisms of action [38]. THC has also been shown to significantly inhibit human P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), which implicates its possible use in reducing resistance to chemotherapeutic agents [39].
2.1.2. Glioma
In glioma cell lines, treatment with THC produced dose-dependent inhibition of cell viability and proliferation [42]. In glioma cells, treatment with THC at 3 µM was able to inhibit cell growth [43]. THC did not have any effects on cell viability in C6 glioma cells when cultured with 10 percent FBS, while it did have modest activity on inhibiting cell viability in a serum-free culture environment [44]. In C6 glioma cells, THC had an IC50 of 23 µM and THC exposure increased cell death as a result of oxidative stress [45]. On the contrary, other studies have shown that human glioma cells were only sensitive to THC at very high, pharmacologically irrelevant concentrations, and that it has the potential to stimulate glioma cell growth [50,128]. An early study by Sánchez and colleagues [46] demonstrated THC’s ability to induce apoptosis in human C6 glioma cells and they suggested that this effect may be mediated through a CB1 receptor-independent pathway involving the stimulation of sphingomyelin breakdown. It has been shown that THC induced autophagy-mediated cell death in glioma cells as a result of ceramide accumulation and endoplasmic reticulum stress [51]. Tribbles homolog 3 (TRB3) linked ER stress to autophagy, and autophagy occurred prior to apoptosis in cannabinoid-induced glioma cell death. This sequence of events was required for the in vivo anti-cancer effects of cannabinoids [51,52]. One study found that long-term exposure to THC did not stimulate apoptosis and actually diminished the sensitivity of astrocytes to ceramide accumulation [129]. Carracedo and colleagues [47] showed that stress protein p8 up-regulation and endoplasmic reticulum stress were necessary for THC-induced apoptosis in glioma cells and cells from human astrocytoma biopsies. In cannabinoid-resistant tumor cells, p8 upregulation did not occur following cannabinoid treatment. They also showed that THC treatment upregulated p8 levels in tumors in vivo, and that tumors deficient in p8 were resistant to the apoptotic effects of cannabinoids. Another study showed that THC treatment modified sphingolipid ratios in the endoplasmic reticulum of glioma cells, which resulted in the promotion of autophagy-dependent lysosomal membrane permeabilization and cathepsin release, resulting in the activation of the mitochondrial apoptotic pathway [48]. Administration of THC also reduced glioma tumor growth in vivo as a result of autophagy-mediated cell death.
THC treatment was able to regress malignant glioma tumors in vivo in murine models. Mice with C6 glioma xenografts were treated for 7 days with THC, which was able to increase survival and reduce tumor progression. THC stimulated apoptosis in glioma cells by accumulation of ceramide and Raf1/extracellular signal-regulated kinase activation [49]. Temozolomide (TMZ) combined with THC synergistically reduced the growth of glioblastoma multiforme xenografts when administered locally [54,55,56]. The combination of TMZ with both THC and CBD together was also looked at, and cannabinoid combinations with higher CBD had a stronger anti-tumor effect in xenografts derived from GBM patients. The same group looked at systemic administration of Sativex-like extracts (1:1 THC:CBD) in combination with TMZ, and anti-tumor effects glioma cell-derived tumor xenografts were still observed. THC resistance can occur in glioma cells, and growth factor midkine (Mdk) has been shown to be involved in this resistance. In vivo, silencing Mdk was able to sensitize resistant tumors to the anti-cancer effects of THC, indicating Mdk as a potential target for improving the effectiveness of cannabinoids in glioma treatment [56]. The local administration of THC downregulated the expression of metalloproteinase-2 (MMP-2) in mice bearing glioma tumors. Likewise, in cultured glioma cells, THC exposure inhibited the expression of MMP-2 and reduced invasion, indicating that reduction in MMP-2 plays a fundamental part in THC-induced reduction of cell invasion. Tissue inhibitors of metalloproteinases, for example MMP-1 (TIMP-1), have been shown to be down-regulated by THC exposure, both in vitro and in vivo, which may explain the THC-induced inhibition of metalloproteinases in glioma cells [53]. THC as a botanical drug substance was more effective than pure THC alone in a murine glioma model [106]. Pre-treatment of glioma cells with pure THC and CBD together increased the sensitivity of glioma cells to radiation therapy both in vitro and in vivo due to increased apoptosis and autophagy [106]. Similarly, another study suggested that adding CBD to the THC treatment of glioblastoma cells may improve the overall efficacy of THC in glioblastoma therapy, as the combination of both cannabinoids had synergistic anti-cancer effects [43]. THC-loaded microparticles for the systemic delivery of cannabinoids have also been developed, and this method of THC delivery facilitated prolonged release for several days, and in mice bearing glioma xenografts the microparticles limited cell proliferation and angiogenesis and increased apoptosis [57].
2.1.3. Leukemia
In a leukemia model, THC had an IC50 of 13 µM and CBD had an IC50 of 8 µM [62]. When THC and CBD were combined in a 1:1 ratio, the IC50 was decreased to 4 µM. They then combined THC/CBD combinations with other anti-cancer agents and in some cases observed synergistic effects, but most importantly, they observed that equivalent anti-cancer effects can still result from lower concentrations of combined agents, compared to each agent alone at a higher concentration. Combinations of THC and CBD also slightly sensitized leukemic cells to anti-cancer agents, vincristine and cytarabine. The beneficial effects of combined therapy with cannabinoids and chemotherapeutic agents was dependent on the sequence of administration; increased cell death was observed when cannabinoids were administered after chemotherapy [62]. THC sensitized leukemia cells to well-established anti-cancer agents, decreasing the IC50 values by approximately 50 percent. The sensitization was found to be due to THC’s ability to down regulate phosphorylated ERK [63]. This data supports the notion that cannabinoids combined with other therapeutic agents may enhance the overall anti-cancer effects. Inhibition of the differentiation blockage in acute myeloid leukemia has been shown to be the most successful target in leukemia therapy. Dronabinol, the enantiomer (−)-trans-Δ9-tetrahydrocannabinol approved by the FDA for conditions like HIV/AIDS-induced anorexia and chemotherapy-induced nausea and vomiting, was found to inhibit the differentiation blockage in acute leukemia cells in vitro and that O-linked-β-N-acetyl glucosamine transferase was fundamental to this process [65].
Dronabinol also reduced cell viability and proliferation, as well as induced apoptosis in an array of acute leukemia cell lines and native leukemic cells cultured ex vivo. Dronabinol’s pro-apoptotic effects in patient-derived leukemic cells correlated with expression of CB1 and CB2 receptors, where the presence of these receptors was necessary to see apoptotic effects. The response to THC treatment was found to be higher in leukemia blasts that were derived from a lymphatic lineage and expressed lymphatic markers [66]. One study suggested that THC-induced apoptosis in leukemia cells occurred as the result of BCL2 associated agonist of cell death (BAD) translocation to the mitochondria. Use of a BAD siRNA was able to reduce THC-induced apoptosis in leukemia cells [67]. Another study used Jurkat leukemia cells with defects in signaling pathways to determine the mechanism of apoptosis induced by THC exposure. The intrinsic pathway was found to play a fundamental role in THC’s ability to induce apoptosis in Jurkat cells [68]. THC also decreased P-gp expression in CEM/VLB(100) cells, which correlated with an increase in the accumulation of P-gp substrate Rh123 and sensitized the cells to vinblastine [64].
2.1.4. Lung Cancer
Low levels of THC induced lung cancer cell proliferation. Metalloprotease and epidermal growth factor receptor (EGFR) activity were found to be fundamental in mediating this increase in cell proliferation [50]. Another study looked at the anti-tumor effects of whole cannabis extracts versus individual compounds alone. In lung cancer cells, they found that treatment with pure THC did not significantly decrease cell survival, relative to control [70]. In contrast, other studies found that THC inhibited epidermal growth factor (EGF) stimulated growth of non-small cell lung cancer and reduced the expression of EGFR, as well as chemotaxis and invasion [71,72]. THC inhibited contact-dependent macrophage cell killing of tumor cells in a cannabinoid-receptor independent manner [73]. Similarly, THC treatment suppressed host immune reactivity to lung cancer and in murine models of lung cancer, administration of THC caused increased tumor growth and decreased tumor immunogenicity [75]. A different study found that THC was able to inhibit tumor growth and lung metastases in a murine model of lung cancer [71]. In non-small cell lung cancer (NSCLC) cells, treatment with THC was able to suppress the epithelial-mesenchymal transition, restore the epithelial phenotype and reduced the proliferation of these cells in vitro. In addition, THC reduced the migration of NSCLC cells [72]. THC-loaded nanoparticles for the treatment of lung cancer caused significant cytotoxicity against human and murine lung cancer cells in vitro and in vivo [74].
2.1.5. Melanoma
Human melanomas have been shown to express both CB1 and CB2 receptors. In melanoma cells, stimulation of these receptors by THC decreased cell viability, proliferation, metastasis, angiogenesis and induced the activation of autophagy and apoptosis [76,78,80]. Treatment with THC was able to diminish the survival of mitogen-activated protein kinase inhibitor (MEKi)-resistant melanoma cells. The combined treatment with a MEK inhibitor (Trametinib) and THC reduced cell viability, invasion and metastasis of MEKi-resistant melanoma cells in vivo [80]. THC has been shown to reduce melanoma cell proliferation and tumor growth in vivo in murine models in other studies [76,79]. In one study, a preparation of equal amounts THC:CBD was able to decrease tumor growth and increase autophagy and apoptosis in vivo [78]. THC significantly inhibited the tumor growth of transplanted mouse melanoma cells in a cannabinoid receptor-dependent fashion [79].
2.1.6. Myeloma
THC has been demonstrated to exert anti-cancer effects in multiple myeloma (MM) cells. THC at concentrations ranging between 30–40 µM was able to inhibit cell viability and proliferation and induce autophagic-dependent necrosis of MM cells. One study found that the combination of THC and CBD had the most potent effects in MM cells compared to each compound on its own. THC and CBD together had synergistic effects with carfilzomib, increasing cell death and decreasing migration. THC treatment alone also reduced migration of MM cells via decreasing the expression of CXCR4 and CD147 (plasma membrane glycoprotein) [77]. Myeloid-derived suppressor cells (MDSC) are induced by cancers with the purpose to evade anti-tumor immune responses and have been shown to be increased in patients with multiple myeloma [81,130]. THC induced MDSCs in mice via epigenetic alterations that promoted MDSC function and differentiation, and S100A8 (a calcium-binding protein) was shown to play a key role in this process [81].
2.1.7. Hepatocellular Carcinoma
THC treatment reduced the viability of hepatocellular carcinoma (HCC) cells in vitro in a CB2-dependent manner. THC also induced autophagy in HCC cells, which was found to be reliant on tribbles homolog 3 (TRB3) up-regulation, and was able to reduce tumor growth in a xenograft murine model [82]. Another study also showed that TRB3 plays a fundamental role in the anti-cancer action of THC, where transformed embryonic fibroblasts derived from TRB3-deficient mice were resistant to the effects of THC [131]. THC increased the activity of PPARγ in HCC cells, and the pharmacological inhibition of PPARγ inhibited the anti-tumor action of THC in these cells, both in vitro and in vivo, indicating that the anti-proliferative actions of THC in HCC cells are influenced by PPARγ-dependent pathways [83]. In a Wistar rat model, co-treatment of irinotecan with THC led to decreased hepatic toxicity, which suggested a protective role of THC on liver injury during acute treatment [85]. In cholangiocarcinoma cells (including a patient sample) THC exposure suppressed proliferation, migration and invasion, and induced apoptosis [84].
2.1.8. Pancreatic Cancer
In pancreatic cancers, cannabinoid receptors have been shown to be much more highly expressed than in regular tissues. Treatment of pancreatic cancer cells with THC was able to decrease cell viability, induce apoptosis, and reduce the growth of tumors in vivo in murine models. The induction of apoptosis by THC in pancreatic cancer cells was the result of ceramide accumulation and endoplasmic reticulum stress, as shown by increased expression of stress protein p8 mRNA [86].
2.1.9. Prostate Cancer
In human prostate cancer cells, treatment with THC was able to reduce cell viability [87]. The IC50 of THC in DU-145 prostate cancer cells was greater than the highest concentration tested (25 µM) [30]. A study found that THC induced apoptosis in a dose-dependent and cannabinoid receptor-independent manner in prostate cancer cells [88].
2.1.10. Colon Cancer
In colorectal cancer cells, THC had an IC50 of 17 µM [30]. Another study found that THC induced apoptosis in colorectal cancer cells via the activation of CB1 receptors and subsequent inhibition of PI3K-AKT and RAS-MAPK/ERK survival pathways [89]. THC biodegradable microspheres were developed for an alternative cannabinoid delivery system than the usual oral route. The THC microspheres were able to inhibit cell proliferation of multiple cancers, including Caco-2 colon cancer cells, over a 9 day period [90].
2.1.11. Endometrial and Cervical Cancers
There are currently very limited options for treatment of aggressive endometrial cancer, and it has been shown that cannabinoid receptors are highly expressed in endometrial cancer tissues. THC treatment of endometrial cancer cells decreased cell viability and motility as a result of inhibiting the epithelial-mesenchymal transition and decreasing metalloproteinase-9 gene expression [132]. ATP-binding cassette (ABC) transporters are highly implicated in the resistance of cancers to anti-cancer drugs. THC was able to increase the accumulation of Fluo3 and Vincristine in ovarian cancer cells that over-expressed ABCC1, both of which are substrates for the transporter [91]. In another study, a different multidrug transporter, ABCG2, was shown to be inhibited by THC, where THC exposure increased the accumulation of mitoxantrone, an ABCG2 substrate [92]. In cervical cancer cells, one study found that THC decreased invasion via the increased expression of tissue inhibitor of MMP-1 (TIMP-1) [93]. This suppression of invasion was reversed by the knockdown of THC-induced TIMP-1 expression.
2.1.12. Oral Cancer
In human oral cancers that are highly resistant to anti-cancer drugs, exposure to THC significantly inhibited mitochondrial oxygen consumption and exhibited strong toxicity in these highly malignant cells [94].
2.1.13. Clinical Results
Inglet et al. [133] recently comprehensively outlined clinical data supporting the use of cannabis-based treatments in a variety of disease states, including cancer. Afrin et al. [27] also presented completed, ongoing, and recruiting clinical trials looking at the effects of cannabis-based treatment in cancer patients. Completed clinical trials that incorporated THC as treatment for various cancers are presented below. One study found that in two patients with pilocytic astrocytomas, tumors regressed over a period of 3 years and neither patient was receiving any conventional adjuvant treatment; however, cannabis was consumed via inhalation over the same period, suggesting cannabis played a role in tumor regression. Unfortunately, no details were available regarding the type, strength, or frequency of cannabis use in these patients [58]. In another study, a patient with terminal acute lymphoblastic leukemia was given Cannabis sativa oil (normally higher in THC content than other cannabinoids), and was able to achieve remission, attributed to the effects of the cannabis oil as the patient was solely on cannabinoid treatment [69]. A two-part clinical study in 2016 investigated the effects of TMZ and Sativex (THC:CBD 1:1) in glioblastoma multiforme patients and found that combination treatment was able to increase the 1 year survival rate by 39 percent (NCT01812603 and NCT01812616). A pilot clinical study looked at the potential of THC treatment in patients with recurrent glioblastoma multiforme. THC inhibited tumor cell proliferation in vitro and reduced tumor cell Ki67 immunostaining when given to 2 patients [59]. In another study, intratumoral injection of THC in two patients suffering from glioblastoma multiforme was able to decrease vascular endothelial growth factor (VEGF) levels, as well as decrease VEGF-2 receptor activation [60]. Dronabinol has had limited use in central nervous system cancers due to the risk of CNS adverse events, however a recent study reported that participants with primary brain tumours did not experience adverse events to the same severity that other studies have reported. This study was however limited by the low number of participants, and the dronabinol dosage used was low (10 mg) relative to other studies [61]. Between 2000 and 2010, pediatric cancer patients receiving chemotherapy were also given low doses of Dronabinol to determine its potential use as an adjuvant antiemetic in children. They found that 60 percent of patients had a positive response to Dronabinol [134]. Dronabinol also inhibited the differentiation blockage in two patients suffering from leukemia [66]. Finally, a study tested the effects of THC in cancer patients in palliative care, and found that daily doses were generally well-tolerated, and nearly 50 percent of patients experienced overall improvement in their well-being [135].
2.2. Cannabidiol (CBD)
Cannabidiol (CBD) is one of the major and most extensively researched phytocannabinoids present in cannabis species. Cannabidiol binds to a large array of physiological targets within the body’s endocannabinoid system. In medical settings, CBD is most commonly administered orally and is commonly prepared as an oil. Cannabidiol is non-intoxicating, which is why its potential as a therapeutic agent is more appealing than some other cannabinoids that do possess psychoactive effects, like THC. To date, there are many studies surrounding the anti-cancer potential of cannabidiol. Two recent reviews by Afrin et al. and Kis et al. [27,136] thoroughly highlighted research studies investigating the anti-cancer effects of cannabidiol. Treatment with CBD exhibited a multitude of beneficial anti-cancer effects in lung, breast, colon, prostate, melanoma, leukemia, cervical, brain, neuroblastoma and multiple myeloma cancer cells (Reviewed in [27,136]), and we highlight these studies here.
2.2.1. Breast Cancer
Several studies have examined the effects of CBD in vitro and in vivo in breast cancer. In breast cancer cells, treatment with CBD has been shown to induce apoptosis and autophagy [30,95]. It has been suggested that CBD can induce endoplasmic reticulum stress apoptosis by enhancing the production of reactive oxygen species (ROS) in select breast cancer cells [95]. Another study demonstrated CBD’s ability to inhibit epidermal growth factor (EGF)-induced proliferation, migration and invasion of breast cancer cells [97]. Recently, CBD’s ability to inhibit the epithelial-mesenchymal transition (EMT) in cancer cells has been an emerging area of research. CBD can revert the EMT in highly invasive breast cancer cells. Treatment of 6D breast cancer cells with CBD was able to significantly reduce migration and invasion, promoted the recovery of cell contacts, and reduced the expression of malignant markers. CBD was also able to increase sensitivity to anticancer agents doxorubicin and cisplatin in 6D cells by down-regulating the expression of resistance proteins [98]. In murine models of breast cancer, CBD reduced cell proliferation and overall tumour growth, as well as migration and invasion to reduce metastasis [97]. One study reported that CBD treatment reduced advanced-stage breast cancer metastasis via the downregulation of Inhibitor of DNA binding protein 1 (Id1), a transcriptional factor [100]. Another study also showed that CBD treatment was able to reduce proliferation and invasion of breast cancer cells via reducing the expression of Id-1. In breast cancer, Id-1 overexpression has been found to be highly correlated with the ability of primary human breast cancer cells to metastasize to the lung [96]. An interesting study by Fraguas-Sanchez et al. [99] looked at the combination of CBD solution or CBD encapsulated in polymeric nanoparticles with chemotherapeutic agents paclitaxel or doxorubicin in breast cancer cells. They found that co-administration of CBD solution or CBD nanoparticles with paclitaxel or doxorubicin had synergistic effects on antiproliferative activity. CBD nanoparticles were also effective as a monotherapy and had prolonged antiproliferative activity, lasting for 10 days, indicating that they may be beneficial for extended release of the cannabinoid during treatment [99].
2.2.2. Lung Cancer
In lung cancer, CBD has been shown to induce apoptosis via cyclooxygenase 2 (COX2) and PPARγ [101]. Several studies using lung cancer cells demonstrated that CBD inhibited invasion and metastasis via decreased secretion of plasminogen activator inhibitor-1 [102,104]. CBD also upregulated the expression of surface protein intercellular adhesion molecule (ICAM-1) in lung cancer cells, which correlated with decreased metastasis of these cells. Additionally, CBD treatment increased the susceptibility of lung cancer cells to adhere to and subsequently be lysed by lymphokine-activated killer (LAK) cells, and that the upregulation of ICAM-1 was responsible for the increased action of LAK cells [105]. One study examined the effects of CBD on the proliferation, migration and EMT in lung cancer cell lines. They found that CBD treatment restored the epithelial phenotype and reduced migration of lung cancer cells [72]. In vivo mouse models of lung cancer showed that treatment with 10 mg/kg/day of CBD resulted in reduced cell viability, decreased overall tumour growth and decreased metastasis [101,103].
2.2.3. Glioma and Neuroblastoma
In gliomas, cannabidiol has been shown to exert anti-cancer effects. In glioma cells, treatment with CBD inhibited cell proliferation and induced apoptosis [43,107,108,109,110]. An interesting study showed that CBD exhibited dose-dependent reduction of cell viability in glioma cells, and that pure CBD was more effective than CBD as a botanical drug substance [106]. A study by Singer et al. [110] showed that CBD-induced apoptosis in glioma stem cells was the result of increased ROS production. An interesting study found that treatment with CBD increased the expression and abundance of heat shock proteins (HSP) in glioma cells as a result of CBD-induced ROS production. Increases in HSP diminished the cytotoxic effects of CBD; when glioma cells were cultured with CBD and HSP inhibitors, the cytotoxic effects were restored. In addition, culturing glioma cells with CBD and HSP inhibitors increased the radio sensitivity of the cells, compared to treatment with CBD alone [112]. In vivo murine models of brain cancer revealed that treatment with CBD was able to inhibit tumor growth, enhance apoptosis and significantly prolong mouse survival [57,108,110]. López-Valero and colleagues [54,55] looked at the combination of Temozolomide (TMZ) with both CBD and THC together in the treatment of glioblastoma multiforme (GBM) in vivo. They found that treatment of glioma cell derived xenografts with TMZ in combination with THC and CBD in a 1:1 ratio and preparations higher in CBD, but not TMZ with CBD alone, exhibited similar anti-tumor effects. On the contrary, in xenografts derived from glioma initiating cells, the combination of TMZ with cannabinoid preparations higher in CBD had stronger anti-tumor effects [54]. The same group looked at systemic administration of Sativex-like extracts (1:1 CBD:THC) in combination with TMZ, and found that treatment was still able to produce anti-tumor effects glioma cell-derived tumor xenografts [55]. In neuroblastoma cell lines, CBD decreased cell growth, induced cell cycle arrest, reduced invasion, and reduced tumor growth in vivo [111]. Another study showed that CBD induced apoptosis in neuroblastoma cells via serotonin and vanilloid receptor activation, and reduced cell migration and invasion in vitro [109]. CBD-loaded microparticles were also used for the treatment of mice bearing xenograft gliomas, where they decreased cell proliferation and angiogenesis of tumors [57].
2.2.4. Myeloma
In multiple myeloma cells, CBD reduced cell viability, increased cytotoxicity of bortezomib and carfilzomib, and inhibited cancer cell migration [77,119].
2.2.5. Colon Cancer
Cannabidiol reduced cell viability, elevated ROS levels and promoted apoptosis in colon cancer cells [113,123]. CBD significantly reduced the number of aberrant crypt foci polyps and tumors in a mouse model of colon cancer. CBD had a chemo-preventative effect on colon cancer cells that was the result of up-regulated caspase-3 [113]. Other in vivo studies demonstrated that treatment with CBD reduced colon cancer cell proliferation, induced apoptosis, and also had anti-metastatic and anti-angiogenesis effects [114]. In HCT116 colon cancer cells, CBD’s antagonistic activity at GPR55 was shown to play a key role in the reduction and prevention of metastasis [117]. In vivo models of colorectal cancer found that CBD treatment induced apoptosis by altering the expression of pro- and anti-apoptotic proteins and decreased overall tumor volume [118].
2.2.6. Prostate Cancer
CBD also exhibits multiple promising anti-cancer effects in prostate cancer studies. Treatment with CBD was able to significantly reduce the growth of various prostate cancer cell lines [87,115]. It was reported by De Petrocellis et al. [87] that CBD inhibited growth of prostate cancer cells via the induction of intrinsic pathways of apoptosis, cell cycle arrest at the G1-S phase and activation p53 and elevated ROS levels. CBD treatment was also able to inhibit tumour growth and increase the effects of bicalutamide and docetaxel in a murine xenograft model [87].
2.2.7. Other Cancers
Cannabidiol has been explored for its potential beneficial effects in melanoma, leukemia, cervical, ovarian and endometrial cancers. In mice injected with melanoma cells, treatment with CBD exhibited very similar effects to treatment with anticancer agent cisplatin, such as increasing survival, significantly reducing melanoma tumour growth, and improving overall quality of life [78,116]. A study by Kalenderglou et al. [137] showed CBD’s ability to decrease cell viability and increase the number of cells in the G1 phase in T acute lymphoblastic leukemia cells. Apoptosis was also induced by CBD exposure in leukemic cells as a result of ceramide accumulation [120]. One study found that CBD decreased P-gp expression in CEM/VLB(100) cells, which correlated with an increase in the accumulation of P-gp substrate Rh123, and sensitized the cells to vinblastine [64]. In cervical cancer cells, treatment with CBD ranging from 1.5 µg/mL to 3.2 µg/mL resulted in the inhibition of cell growth and apoptosis [122]. ABC transporters are highly implicated in the resistance of cancers to anti-cancer drugs. In ovarian cancer cells over-expressing ABCC1, CBD exposure was able to increase the intracellular accumulation of 2 ABCC1 substrates, Fluo3 and Vincristine [91]. Another study showed that CBD inhibited multidrug transporter ABCG2 and promoted the intracellular accumulation of mitoxantrone, a substrate for this transporter [92]. In endometrial cancer, concentrations of CBD higher than 5 µM significantly reduced cell viability. CBD increased levels of caspase 3/7, reactive oxygen species and cleaved poly (ADP-ribose) polymerase (PARP) in Ishikawa cells, indicating induction of apoptosis. The activation of transient receptor potential cation channel subfamily V member 1 (TRPV1) was fundamental in facilitating CBD’s anti-cancer effects in endometrial cancer cells [121].
2.2.8. Clinical Results
There have been a few clinical trials involving multiple cancer types that looked at the therapeutic potential of CBD treatment in cancer patients. Afrin et al. [27] highlighted completed, ongoing and recruiting clinical trials looking at effects of cannabinoids in cancer, some of which included CBD treatment. The completed trials that included CBD as treatment are outlined here. In 2016, a two-part clinical trial (NCT01812603 and NCT01812616) was performed to assess the effectiveness of Sativex (1:1 THC:CBD) in combination with Temozolomide in glioblastoma multiforme patients, and found that it was able to increase 1 year survival rate by 39 percent. Another study tested the effects of CBD in cancer patients undergoing palliative care, and found that daily doses of CBD were generally well-tolerated, and 50 percent of patients experienced overall improvement in their condition [135]. They did, however, mention that these results need to be replicated in a trial with placebo controls.
2.3. Cannabigerol (CBG)
Cannabigerol (CBG) is one of the main active phytocannabinoids produced by Cannabis Sativa L. plants; however, it is considered a minor phytocannabinoid due to its lower abundance relative to THC and CBD. Cannabis cultivars that tend to have higher cannabigerol content are referred to as Type IV cannabis [138]. Cannabigerol is derived from its acidic precursor cannabigerolic acid (CBGA), which also serves as the precursor molecule for the production of THC and CBD. Recently, cannabigerol has attracted more attention for its use in therapeutics due to its lack of intoxicating effects, and more commercial hemp varieties have been developed with CBG and CBGA as the main phytocannabinoids present [139]. As of yet, only a handful of studies have been done to investigate the anti-cancer potential of cannabigerol. Two early studies by Baek et al. [124,125] looked at the potential therapeutic benefits of cannabigerol in mouse skin melanoma cells and oral epithelioid carcinoma (KB) cells. In mouse skin melanoma cells, CBG was found to have significant inhibitory effects on proliferation, with an IC50 of 3 µg/mL. In KB cell lines, CBG over a concentration range of 1–100 µM was the most effective of the cannabinoids tested at reducing cell viability. In MDA-MB-231 breast carcinoma cells, 25 µM CBG was shown to inhibit the uptake of [14C]anandamide and activate vanilloid receptor TRPV1 [30]. CBG also stimulated apoptosis, ROS production, up-regulated C/EBP homologous protein (CHOP) mRNA and inhibited cell proliferation in colorectal cancer (CRC) cells. In vivo, CBG was shown to decrease the growth of xenograft tumours in a murine model, and that this effect was largely due to its activity as an antagonist at TRPM8 receptors on CRC cells [123].
2.4. Cannabichromene (CBC)
Cannabichromene is considered another one of the minor phytocannabinoids produced by Cannabis sativa L. plants, due to its lower abundance than the major cannabinoids THC and CBD. In the United States, cannabichromene has been found to be the second most abundant type of cannabinoid present in some strains of cannabis, particularly abundant in dry-type cannabis material. Though CBC is commonly found in many cannabis strains, there is much to be discovered about its pharmacology. Like CBD, cannabichromene lacks intoxicating effects and is therefore appealing to researchers in terms of its potential therapeutic effects in human health and medicine [140]. The potential anti-cancer effects of cannabichromene have not been extensively studied. In prostate carcinoma (PCC) cells DU-145 and LNCaP, cannabichromene was found to be the second most potent inhibitor of cell viability behind CBD, and CBC at 10 µM had very little effect on caspase 3/7 activity. In serum deprived PCC 22RV1 and PC-3 cells, treatment with CBC at 20 µM resulted in significant activation of caspase 3/7, and CBC caused elevated intracellular Ca2+ in all four PCC cell lines mentioned [87]. In colorectal cancer Caco-2 cells, CBC was able to inhibit cell growth, but only at a concentration of 30 µM [123]. In MDA-MB-231 and MCF-7 breast cancer cell lines, CBC demonstrated high potency as an inhibitor of cell viability [30].
2.5. Cannabidivarin (CBDV)
Cannabidivarin’s structure is similar to cannabidiol except with a shortened side chain. Cannabis cultivars with relatively high levels of CBDV have been identified in India and Nepal. In one study, CBDV was assessed for potential cytotoxic effects on various human prostate carcinoma cell lines. Results showed an IC50 of around 20 µM [87]. In colon cancer cells, CBDV inhibited cell viability in a concentration-dependent manner, with an IC50 of 10 µM [123].
2.6. Cannabinol (CBN)
Cannabinol (CBN) is present in the cannabis plant and, particularly in aged cannabis, is the degraded product of tetrahydrocannabinolic acid. While it was the first of the phytocannabinoids to be isolated, it remains poorly studied. Cannabinol’s psychoactive effects are estimated to be 10 times lower than that of THC [141]. Some cytotoxic effects were observed for cannabinol in prostate cancer cell lines DU-145 and LNCaP. The observed IC50 was reported to be superior to the highest dose tested (25 µM) in most experiments [87]. CBN has also been shown to have some antiproliferative effects in aggressive breast cancer cells [96]. Multi-drug transporters are an ongoing issue in the treatment of cancers due to their ability to confer resistance to multiple anti-cancer agents. In one study, CBN inhibited multidrug transporter ABCG2 and promoted the accumulation of mitoxantrone, a substrate for this transporter [92].
2.7. Cannabivarin (CBV)
Cannabivarin (CBV), also known as cannabivarol, is found in minor amounts in some cannabis cultivars, and it is an analog of cannabinol with a shortened side chain. It is considered an oxidization product of tetrahydrocannabivarin and is rarely found in fresh cannabis. There does not appear to be any published literature surrounding the biological effects of cannabivarin (or cannabivarol) in cancer.
2.8. Tetrahydrocannabivarin (THCV)
Tetrahydrocannabivarin (THCV) is a homologue of THC, where different side chains contribute to a variety of effects that are distinct from THC. Most cannabis cultivars only contain trace amounts of THCV, but some sativa cultivars from hybridized African genetics may have higher levels of THCV. As for most other minor cannabinoids, little has been demonstrated regarding the effects of THCV in cancer. Some cytotoxic effects were observed for tetrahydrocannabivarin in prostate cancer cell lines DU-145 and LNCaP, with IC50 values above 17.5 µM [87].
3. Terpenes
More than 20,000 terpenes appear in nature, from every plant, flower, and even some insects. Relatively few of these compounds–about 200–are found in cannabis. According to recent publications [142,143], 50 cannabis terpenes can be found in North American chemovars, but some are more commonly found (Figure 2). The monoterpene myrcene as well as the sesquiterpenes β-caryophyllene and α-humulene appear to be present in most cannabis cultivars. Other compounds commonly found include alpha-pinene, limonene, linalool, bisabolol and (E)-β-farnesene while some others, in particular sesquiterpenes, are difficult to identify. As a result, the reported terpene profiles of cannabis cultivars may present incomplete portraits of the actual terpenes present in the plant [144]. Furthermore, even within a plant, the localization of the sample taken may also alter the terpene profile. Stereochemistry is also not consistently described in cannabis cultivars. These issues make it difficult to fully understand the diversity of terpenes in cannabis and complicates the analysis of studies using extracts or botanical preparations [143]. Generally, terpenes are typically found in cannabis flowers at levels of 2–5%, but can have much higher concentrations in various products (vaping oils, for example). Yet, information about many of the terpenes is available in regard to their potential beneficial effects. Some of those effects, related to cancer, are described below and in Table 2.
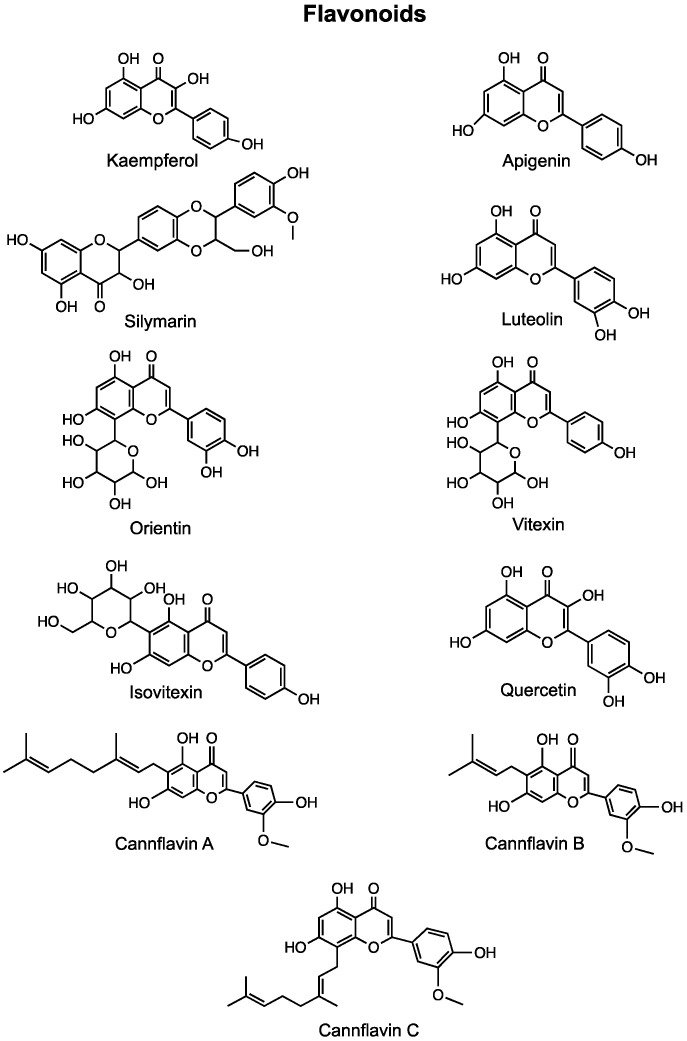
Table 3.
Anti-Cancer Effects of Flavonoids Present in Cannabis.
| Compound | In Vitro Effects | In Vivo Effects | Clinical Trials |
|---|---|---|---|
| Kaempferol | Inhibited cell viability in a dose-dependent manner [327,328,329,330,331,332,333,334,335]. Induced cell cycle arrest at the G2/M or G0/G1 phase [329,335,336,337,338,339,340]. Reduced migration and invasion [340,341,342,343,344,345,346]. Inhibited the EMT and reduced resistance to chemotherapeutic agents [343,347]. Altered expression of VEGF [348]. Induced apoptosis and autophagy [330,340]. Kaempferol + Luteolin inhibited cell proliferation, induced cell death, inhibited migration and invasion [349]. Kaempferol + TRAIL induced apoptosis [350]. Kaempferol + 5-fluorouracil had synergistic anti-proliferative effects and re-sensitized resistant cells to chemotherapeutic agents [341,351]. |
Increased mouse survival [339,352,353,354,355]. Reduced tumor growth and metastasis [339,352,353,354,355,356]. Caused degranulation and accumulation of mediators in leukemia cells [357,358]. |
N/A |
| Apigenin | Reduced cell viability and proliferation [359,360,361]. Induced cell cycle arrest at the G1 or G2/M phase [360,362,363]. Inhibited hypoxia-induced resistance via suppression of HIF-1α [362]. Enhanced activity of paclitaxel [362]. Apigenin + Sorafenib increased apoptosis and decreased migration and invasion [364]. Apigenin + Abivertinib had synergistic anti-cancer effects [360]. Induced apoptosis and reduced angiogenesis [361]. |
Exacerbated the effects of paclitaxel [362]. Inhibited tumor growth via ER-mediated PI3K/Akt/mTOR pathway [365]. Apigenin + Abivertinib exhibited synergistic anti-cancer effects [360]. Apigenin combined with IL-6 inhibition potentiated anti-cancer effects of apigenin [365]. |
N/A |
| Cannflavin B | Increased apoptosis [366]. | Delayed local and metastatic tumor progression [366]. Increased survival [366]. |
N/A |
| Silymarin | Induced apoptosis [367,368,369,370]. Reduced cell viability and proliferation [368,370]. Silymarin nanoemulsion + cold atmospheric plasma reduced intracellular ATP levels and down-regulate transcriptional and survival pathways [371]. Inhibited EMT and migration [372]. |
Reduced tumor volume and induced apoptosis [369]. | High dose silibinin was well tolerated in patients; common adverse event observed was asymptomatic liver toxicity [373]. |
| Luteolin | Caused cell cycle arrest [362,374,375,376,377]. Decreased cell viability and proliferation [378,379,380,381,382,383]. Inhibited the EMT [376,381,384]. Inflicted double-stranded DNA breaks and prevented nonhomologous end joining [385]. Induced apoptosis [374,375,382,386,387,388]. Reduced migration and invasion [378,379,388,389,390]. Luteolin + Oxaliplatin inhibited proliferation, induced apoptosis and altered the cell cycle [391]. |
Inhibited cell growth [378]. Reduced migration, invasion and metastasis [384]. Inhibited angiogenesis [387]. Decreased tumor volume and dimension [377]. |
N/A |
| Orientin | Reduced migration and invasion [392]. Induced apoptosis and altered apoptotic protein levels [393,394]. Caused cell cycle arrest [394,395]. Decreased cell proliferation [393,395]. |
Antiproliferative effects [396]. Improved tumor marker levels and decreased proliferative marker levels [396]. Reduced occurrence of polyps and aberrant crypt foci [397]. Increased antioxidant defense [397]. |
N/A |
| Vitexin & Isovitexin | Reduced cell viability and proliferation [398,399,400,401]. Induced apoptosis [398,399,401,402,403,404]. Caused cell cycle arrest at the G2/M phase [399]. Vitexin + 5-fluorouracil had synergistic anti-tumor effects via PUMA induction [402]. Vitexin + Doxorubicin + Sorafenib induced apoptosis [405]. |
Inhibited cell/tumor growth [399,401,403,406]. Induced apoptosis [403]. Reduced overall tumor size [401]. |
N/A |
| Quercetin | Decreased cell viability and proliferation [407,408,409,410]. Induced apoptosis [407,409,411,412]. Reduced migration [413]. Increased the radiosensitivity of cells [414]. Reversed docetaxel resistance [411,415]. Inhibited the EMT and downregulated expression of MALAT1 [412]. Quercetin + Paclitaxel reduced cell proliferation, migration, and induced apoptosis and cell cycle arrest [416]. Quercetin + Doxorubicin caused increased cytotoxicity and induced apoptosis [417,418]. Quercetin + Gemcitabine caused increased apoptosis in gemcitabine-resistant cancer cells [419]. |
Inhibited cell proliferation and tumor growth [408,411,412]. Delayed appearance of lung adenocarcinoma [420]. Reversed docetaxel resistance [411]. Inhibited breast cancer resistance protein [421]. Quercetin + Paclitaxel increased anti-cancer effects of paclitaxel [416]. Quercetin + Doxorubicin decreased tumor growth [418]. Quercetin + Docetaxel decreased tumor growth [411]. |
4.1. Kaempferol
Kaempferol is a well-characterized natural flavonol that is commonly found in dietary items like tea, apples, strawberries, broccoli, and beans [327]. It is also produced by the Cannabis plant and has attracted much research surrounding its potential health benefits, including its potential as an anti-cancer agent. A plethora of research in recent years has demonstrated many of kaempferol’s anti-cancer effects in vitro and in vivo on a variety of cancer subtypes. Two comprehensive reviews by Irman et al. [328] and Kashyap et al. [327] discussed the anti-cancer effects of kaempferol in various cancers. Here we highlight the main review findings and discuss more recent studies that looked at kaempferol’s potential as an anti-cancer agent. Kaempferol treatment inhibited cell viability in a dose-dependent manner in a multitude of cancer subtypes. Most of the studies reviewed indicated that kaempferol’s inhibitory effects on cell viability in cancer cells were as a result of cell cycle arrest or apoptosis. Kaempferol was able to induce cell cycle arrest at the G2/M phase in a multiple cancers, including leukemia, breast, liver, stomach and ovarian cancers [329,336,337,338]. In oral cancers, kaempferol also induced cell cycle arrest, but alternatively at the G0/G1 phase [339]. In glioblastoma, hepatic, colorectal, pancreatic, lung, renal and breast cancer cell lines, kaempferol was able to significantly reduce migration and/or invasion in vitro [341,342,343,344,345]. In lung and breast cancer cell lines (A549 and MDA-MB-231/MCF-7), studies have found that treatment with kaempferol was able to inhibit the epithelial-mesenchymal-transition (EMT), resulting in decreased metastasis and resistance to chemotherapeutics in these cells [343,347]. The detailed mechanisms of the anti-cancer actions of kaempferol in breast cancer can be found thoroughly reviewed by Wang et al. [422]. Several studies have demonstrated the ability of kaempferol to reduce angiogenesis; this has been demonstrated in ovarian cancer cells as a result of altered expression of vascular endothelial growth factor [348]. Studies investigating the anti-cancer effects of kaempferol in vivo were also reviewed by Irman et al. [328,423]. In in vivo mouse models of various cancers, including bladder, oral, prostate, lung and bone cancers, treatment with kaempferol was able to increase survival and reduce the growth and metastasis of tumours [339,352,353,354,355]. In a rat model of leukemia, treatment with kaempferol resulted in degranulation in basophilic leukemia cells (RBL-2H3) and increased the accumulation of mediators in human leukemic mast cells (HMC-1) [357,358].
In addition to the studies highlighted in the reviews mentioned previously, several studies have since been published that investigated the anti-cancer potential of kaempferol. In gastric, colon, prostate, colorectal and neuroblastoma cancer cells, kaempferol was able to significantly decreased cell viability and proliferation [330,331,332,333,334]. Kaempferol treatment induced autophagy in gastric cancer cells [330]. In breast cancer cells, kaempferol was able to induce apoptosis, cell cycle arrest at the G2/M phase, and suppress cell proliferation [329]. In a mouse model of breast cancer, kaempferol treatment was able to suppress primary tumour growth and lung metastasis [356]. In ovarian cancer cell lines, treatment with kaempferol inhibited growth with IC50 values between 25–50 µM [335]. Further investigation revealed that kaempferol caused cell cycle arrest at the G2/M phase and induced apoptosis in OVACAR-3 ovarian cancer cells due to upregulation of apoptotic proteins such as caspase 3 and Bax [335]. Kaempferol exhibited anti-proliferative effects in endometrial carcinoma cells through apoptosis and cell cycle arrest, and was able to decrease migration and invasion trends in these cells [340]. In immortalized human retinal pigment epithelial (ARPE-19) cells, treatment with kaempferol was able to decrease cell migration through ERK1/2 signaling [346]. Kaempferol-conjugated gold nanoclusters (K-AuNCs) were developed by Govindaraju et al. [424] as a potential anti-cancer drug delivery system, and they demonstrated that K-AuNCs targeted A549 lung cancer cells and exhibited toxicity via nucleus damage.
A few studies have investigated the potential of combination treatment with kaempferol and previously established anti-cancer agents or other compounds. A study by Seydi et al. [349] using cancerous hepatocytes from a rat model of hepatocellular carcinoma found that kaempferol combined to luteolin (another common flavonoid) was able to inhibit cell proliferation, induce cell death and inhibit migration and invasion. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) stimulates apoptosis through binding death receptors 4 and 5 in a variety of cancers, however resistance to TRAIL has been known to occur [350]. Hassanzadeh et al. [350] found that co-treatment of lymphoblastic leukemia (MOLT-4) cells with TRAIL and kaempferol was able to induce apoptosis by inhibiting the expression of anti-apoptotic proteins and up-regulation of death receptors 4 and 5, and they suggested that this co-treatment could be used as a potential solution to overcome resistance to TRAIL in cancers. Li et al. [351] looked at the potential of combining 5-fluorouracil (5-FU) with kaempferol in colorectal cancer. They found that the combination of 5-FU and kaempferol was superior at inhibiting cell viability than either agent alone, and the anti-cancer effects were mediated through reduction in cell proliferation and induction of apoptosis. Another study looked at the combination of 5-FU with kaempferol in 5-FU-resistant colon cancer cells [341]. They found that combination treatment of 5-FU with kaempferol had synergistic effects on cell viability and was able to chemo-sensitize the resistant cells.
4.2. Apigenin
Apigenin is a natural flavone found in many fruits and vegetables, and predominantly found in parsley, celery, and in the flower of chamomile plants, among others. Apigenin is a pigment, yellow in color. A comprehensive review by Imran et al. [425] provides details about the anticancer effects of apigenin in various types of cancer such as breast, lung, liver, brain, skin, blood, bone, colon, prostate, pancreatic, cervical, ovarian, oral, and stomach. Detailed mechanisms of action of apigenin in each of these cancer types can be found within the review however, the induction of apoptosis, upregulation of caspases-3, -8 and TNF-α, downregulation of MMP-2, -9, NFkB, PI3K, Akt and pAkt, and modulation of kinases are mechanisms frequently involved. Since the review’s publication, further studies have investigated apigenin’s anticancer effects. Apigenin reduced cell viability in MCF-7, A549, HepG2 and normal HEK 293 cell lines with the greatest activity against HepG2 liver cancer cells (EC50 of 12 µg/mL) [359]. Li et al. [362] further noted that in three hepatocellular carcinoma cell lines, apigenin was cytotoxic and induced G1 phase cell cycle arrest in a dose dependent manner through the regulation of CyclinD1 and CDK4. Additionally, hypoxia-inducible factor 1α (HIF-1α) is associated with hypoxia-induced resistance in cancer cells but has been shown to be inhibited by apigenin. Two pathways involved in suppressing the HIF-1α expression in hypoxic tumors are through the inhibition of the AKT/p-AKT pathway and HSP90, which also enhance the activity of the chemotherapeutic paclitaxel. Apigenin and paclitaxel also acted synergistically in a liver cancer cell line and murine models [362]. When combined with the chemotherapeutic sorafenib, apigenin decreased cell viability of liver cancer cells to a greater extent than either drug alone. This combination caused an increase in apoptosis and decreased the migration and invasion capability of the cells [364].
In a colon cancer and lymph-endothelial cell model, treatment with apigenin reduced the formation of circular chemorepellent-induced defects in the endothelial barriers [426]. In a cervical cancer model, apigenin inhibited cell growth in vitro and tumour growth in vivo through the ER-mediated PI3K/Akt/mTOR pathway [365]. Similarly, in a model of diffuse large B-cell lymphoma, apigenin inhibited proliferation and colony formation by activating pro-apoptotic proteins, downregulating cell cycle proteins to increase G2/M phase arrest and inhibiting the PI3K/mTOR pathway. Apigenin also acted synergistically in vitro and in vivo with Abivertinib, a bruton tyrosine kinase inhibitor, which can provide new options for patients who have developed resistance to traditional therapies [360]. Apigenin altered the tumor necrosis factor and IL-10 release by microglia. When treated with conditioned medium of microglia treated with apigenin, C6 glioma cells exhibited reduced tumor migration and viability, due to the reduction in IL-6 levels. Apigenin also preferentially reduced viability of C6 glioma cells when co-cultured with microglia [363]. One study showed that apigenin inhibited cell proliferation, induced apoptosis, reduced vascular endothelial growth factor (VEGF) expression, and reduced tumor-induced angiogenesis in two human esophageal cancer models [361]. The inhibition of IL-6 transcription further potentiated these effects, suggesting that the inhibition of IL-6 transcription was how apigenin exhibited its anticancer effects in esophageal cancer cells. Similar effects were seen in a murine xenograft model [365].
4.3. Cannflavins
Cannflavins are a group of prenylflavonoids uniquely found in cannabis. Cannflavins A and B are formed by a derivative of luteolin, chrysoeriol [427]. Recently, a study examined the potential of cannflavin B derivatives for the treatment of pancreatic cancer. In vitro results showed an increase in apoptosis in two pancreatic cancer cell lines treated with concentrations of FBL-03G (or caflanone), the cannflavin B derivative. In vivo local and metastatic tumor progression were delayed in pancreatic cancer animal models as well as an increase in survival compared to control cohorts [366]. In 2019, caflanone was granted orphan drug status by the United States Food and Drug Administration and clinical trials with the drug were scheduled to begin as potential treatment for pancreatic cancer. Caflanone has been identified in a rare, flavonoid-rich cannabis cultivar native to Jamaica known as Black Swan. Little is known about the potential actions of Cannflavin A and Cannflavin C in cancer.
4.4. Silymarin
Silymarin is a flavonoid derived from milk thistle, but is also present in artichokes, cilantro, coriander, and cannabis. Silymarin consists of three phytochemicals, silybin, silidianin, and silicristin, and has a long medicinal tradition. Silybin is its most active phytochemical and is largely responsible for the effects of silymarin. A recent review by Delmas et al. [428] provided extensive details about mechanistic actions of silymarin in various models of cancer. The review highlighted silymarin’s ability to synergize with anticancer drugs, induce cell death through both the intrinsic and extrinsic pathways, cause cell cycle arrest in the G0/G1 and G2/M phase, modulate metabolizing enzymes and drug transporters which alter cellular sensitivity to chemotherapeutics, as well as several clinical trials currently in progress. More recently, in a Burkkett’s lymphoma model, silymarin induced apoptosis and caused a reduction in toll-like receptor 8 (TLR8) mRNA expression, implicating toll-like receptors in silymarin’s anticancer activity [367]. Silymarin further decreased cell viability, increased apoptosis, and changed the mitochondrial membrane potential in glioblastoma cells [368]. Silymarin reduced cell viability and diminished migration of stomach cancer cells through the induction of apoptosis, inhibition of p-ERK and activation of p-p38 and p-JNK. In vivo, silymarin at a concentration of 100 mg/kg reduced tumor volume and induced apoptosis [369]. Additionally, silymarin dose-dependently inhibited cell growth in prostate cancer cells by initiating apoptosis. After treatment, the expression of Slit Guidance Ligand 2 (SLIT2) and Roundabout Guidance Receptor 1 (ROBO1) were increased and the expression of CXCR4 was decreased [429]. Silymarin also had antiproliferative, antimetastatic and pro-apoptotic effects in a dose-dependent manner on liver cancer cells, and also acted through the Slit-2/Robo-1 pathway [370]. Co-treatment with cold atmospheric plasma (CAP) and a silymarin nanoemulsion (SN) decreased intracellular ATP levels and downregulated the PI3K/AKT/mTOR survival and RAS/MEK transcriptional pathways in melanoma cells [371]. Silibinin (30–90 μM), a main active component of silymarin, inhibited the epithelial-mesenchymal transition (EMT) in breast cancer cells. Silibinin also inhibited cell migration and increased mitochondrial fusion, which contributed to silibinin’s inhibitory effect on cell migration. Additionally, silibinin decreased ROS production, which decreased the NLRP3 inflammasome activation [372]. In silico, silymarin was shown to inhibit the proto-oncogene B-Raf (BRAF) and the smoothened gene (SMO), two targets in anticancer therapy [430]. A phase I clinical trial studied the effects of high dose silibinin (13 g daily) and found it to be well tolerated in patients with advanced prostate cancer. The most commonly seen adverse event was asymptomatic liver toxicity (hyperbilirubinemia and elevation of alanine aminotransferase) [373].
4.5. Luteolin
Luteolin is a flavone commonly found in several plants including broccoli, pepper, thyme, and celery. Luteolin has been used as a source of yellow dye since at least the first millennium B.C. and originally obtained from the plant Reseda luteola, a common weed. A recent review by Imran et al. [328] provides extensive information about the anticancer effects of luteolin in many cancers including breast, prostate, oral, lung, kidney, cervical, placental, ovarian, skin, liver, esophageal, bladder and glioblastoma. This review provides insight about mechanisms involved in luteolin’s anticancer effects ultimately leading to reductions in cell proliferation, cell survival signaling, angiogenesis, and metastasis and an increase in apoptosis in many of these types of cancers [431]. Since this review’s publication, more studies have further evaluated luteolin’s anticancer effects.
In vivo models of melanoma showed that luteolin inhibited cell growth through the extracellular matrix pathways, the oncogenic signaling pathway, and the immune response pathways, but not through ROS induction [378]. Furthermore, luteolin reduced proliferation, migration, invasion, adhesiveness, and tube forming potential in a metastatic melanoma model. HIF-1α/VEGF signaling-mediated epithelial to mesenchymal transition and angiogenesis was implicated in the anti-metastatic effects demonstrated by luteolin [379]. Luteolin caused double-strand DNA breaks and prevented nonhomologous end joining (NHEJ) and homologous recombination (HR) in a bursal lymphoma model, and additionally caused G2/M phase cell cycle arrest in BRCA-deficient cells and inhibited Poly [ADP-ribose] polymerase 1 (PARP1) [362,385].
Luteolin decreased cell viability in breast cancer cells as well as inhibited migration and invasion by decreasing the expression of matrix metalloproteinase-9 (MMP9) [378,379,432]. Apoptosis was induced through the extrinsic and intrinsic pathways and the epithelial-mesenchymal transition (EMT) was prevented. This was mediated by increased expression of miR-203, reduced Ras/Raf/MEK/ERK signaling, cell cycle arrest at the S phase, and by reducing telomerase levels through suppressing human telomerase reverse transcriptase (hTERT) expression [374]. S100 calcium-binding protein A7 (S100A7) has been implicated in the EMT, promoting metastasis, and was inhibited by luteolin through Src/Stat3 signaling in epidermoid carcinoma cells. This reduced migration and invasion of A431-III cells and decreased metastasis in a xenograft zebrafish model [384]. Similarly, miRNA-301-3p was downregulated in pancreatic cancer cells following treatment with luteolin, causing a decrease in cell growth [380]. In several ovarian cancer cell lines, luteolin induced apoptosis through the extrinsic and intrinsic pathways. The cell cycle was disrupted and cell invasion on the collagen was altered [375]. In non-small cell lung carcinoma, luteolin induced G2/M cell cycle arrest and reduced EMT by reducing the expression of absent in melanoma 2 (AIM2), leading to decreased AIM2 inflammasome activation which was also seen in lung cancer mouse xenograft models [376]. Masraksa et al. [389] reported that luteolin showed no cytotoxic activity on lung cancer cells up to 40 µM, however 20–40 µM was able to reduce migration, invasion and the formation of filopodia in a concentration dependent manner.
Luteolin has also had various anticancer effects in colon cancer cells. Luteolin reduced the viability and proliferation of colon cancer cells and increased the expression of pro-apoptotic and pro-autophagic proteins in a concentration-dependent manner. The EMT process was reversed after treatment, and the ERK/FOXO3a-dependant mechanism was implicated in these anticancer effects [381]. Luteolin reduced cell viability by inducing apoptosis and this effect was increased in p53-expressing cells. Treatment also reduced oxaliplatin-treated p53-null cell viability and colony counts further and may provide a new treatment for colon cancer cells resistant to oxaliplatin [386]. Luteolin increased the expression of pro-apoptotic proteins and antioxidant enzymes in colon cancer cells [433]. Treatment increased the number of sub-G1 phase cells and cells with fragmented nuclei and increased the expression of the Nrf2 promoter and altered its interaction with the tumor suppressor p53 [433]. Interestingly, one study found that luteolin did not impact cell proliferation of colorectal cancer in vitro or in vivo; however, it inhibited cell migration and invasion. The downregulation of pleiotrophin (PTN) via miR-384 was implicated in luteolin’s effects [390]. In liver cancer cells, luteolin reduced NF-κB transcription factor activation and decreased COX-2 gene expression. It also promoted cytotoxic effects including inhibition of proliferation, ER stress, and induction of apoptosis in a model lacking p53 [382].
The serine-threonine kinase CK2–overexpressed in all cancers where it promotes proliferation, spread, and survival–is inhibited by luteolin [383]. Luteolin induced apoptosis and inhibited the progression of rat prostate carcinogenesis in a transgenic rat for adenocarcinoma of prostate (TRAP) model in addition to a xenograft prostate cancer model where angiogenesis was also inhibited. In another human and rat cell model of prostate cancer, luteolin induced apoptosis by activation of caspases-3 and 7 [387]. Additionally, migration and tumorigenesis were inhibited, and apoptosis was induced in a glioblastoma model. Apoptosis was caused by depolarization of the mitochondrial membrane, ERK protein phosphorylation, cleavage of PARP and caspase-9, causing DNA damage by H2AX phosphorylation [388]. Bladder cancer cell survival was inhibited by luteolin by inducing G2/M cell cycle arrest, p21 upregulation and inhibition of mTOR signalling. In xenograft models, tumor volumes and dimensions were decreased after oral administration of luteolin [377]. When combined with the anticancer agent oxaliplatin, luteolin inhibited gastric cancer cell proliferation and induced apoptosis through the cytochrome-c and caspase pathways and by altering the cell cycle [391].
4.6. Orientin
Orientin is a flavone and glucoside derivative of luteolin. Orientin is found in various plants and flowers including the passion flower, bamboo leaves, açaí palm, buckwheat sprouts and in millets. Orientin had anti-migratory and anti-invasive effects in TPA-treated MCF-7 cells. Orientin downregulated TPA-induced membrane translocation of protein kinase C-α, phosphorylation of extracellular signal regulated kinase, and nuclear translocations of activator protein-1 and signal transducer and activator of transcription 3 and inhibited matrix metalloproteinase-9 and IL-8 expression. This resulted in reduced migration and invasion of the MCF-7 cells [392]. Similar inhibitory effects were shown in EC-109 esophageal squamous carcinoma cells in a time and concentration-dependent manner. Orientin-induced apoptosis occurred as a result of upregulated p53 and downregulated pro-apoptotic protein Bcl-2 [393]. Orientin also had cytotoxic and antiproliferative effects against human colorectal cancer HT29 cells. In these cells, orientin induced apoptosis and cell cycle arrest at the G0/G1 phase through the regulation of cyclin and cyclin-dependent protein kinases [394]. Additionally, orientin inhibited T24 human transitional cell bladder carcinoma cell proliferation, induced cell cycle arrest, and decreased the expression of inflammatory mediators such as NF-ĸB and components of the Hedgehog signaling pathway, at a concentration of 100 µM [395]. In vivo, orientin administration (10 mg/kg) resulted in antiproliferative effects on 1,2-dimethyl hydrazine (DMH)-induced colorectal cancer in rats, and improved tumor marker levels while decreasing proliferative marker levels, such as proliferating cell nuclear antigen (PCNA) and Ki67 [396]. Orientin also reduced the occurrence of colonic polyps and aberrant crypt foci and increased the antioxidant defense in rats with DMH-induced colorectal cancer, demonstrating its antiproliferative and antioxidant properties in vivo [397].
4.7. Vitexin and Isovitexin
Vitexin and Isovitexin are apigenin flavone glucoside, a chemical compound formed by the combination of apigenin with sugars found in the passion flower, chasteberry, and some bamboo leaves, among other plants. Isovitexin is also known as homovitexin or saponaretin. Anticancer effects have been observed for both these compounds. A review by Ganesan and Xu [434] provides insight into vitexin and isovitexin’s anticancer effects in various in vitro and in vivo models of cancer. The review highlights vitexin and isovitexin’s ability to inhibit cell growth, induce apoptosis, reduce autophagy, reduce cell migration, and provides further mechanistic details about the specific pathways involved. More recently, vitexin has been shown to reduce the viability of adenocarcinomic human alveolar basal epithelial cells dose-dependently, with virtually no toxicity to normal human bronchial epithelial cells. Vitexin induced apoptosis in these cells by increasing pro-apoptotic protein expression, reducing mitochondrial membrane potential and through Akt signalling [398]. In a melanoma model, vitexin inhibited growth in vitro and in vivo, arrested the cell cycle in G2/M phase, induced apoptosis, caused DNA damage, and increased ROS accumulation. Vitexin increased the ROS levels, causing DNA cytotoxicity leading to G2/M cell cycle arrest and apoptosis in BRAFi (BRAF inhibitor)-resistant melanoma cells [399]. Vitexin has shown anticancer effects in several colon cancer models. Chen et al. [402] showed that vitexin promoted apoptosis through p53, p53 upregulated modulator of apoptosis (PUMA) and Bax (Bcl-2-associated X protein) activation. When vitexin was combined with the anticancer agent 5-fluorouracil, a synergistic antitumor effect via PUMA induction was observed. In a multidrug resistant human, colon cancer cell line vitexin had cytotoxic effects by inhibiting autophagy and inducing apoptosis through decreasing autophagy related protein expression and increasing pro-apoptotic protein expression. Additionally, it induced apoptosis and suppressed tumor growth in a multidrug resistant human colon cancer xenograft model [403]. It was also noted that heat shock factor 1 (HSF-1) is a potential target of vitexin in this multidrug resistant model [435]. Both vitexin and isovitexin exhibited anticancer effects in a liver cancer model. These effects were achieved via blocking the STAT3 signaling cascade and reducing survival and invasion. When combined with doxorubicin and sorafenib, vitexin had apoptotic effects [405]. Isovitexin had antiproliferative effects in prostate cancer cells and osteosarcoma cells [400,401]. In the osteosarcoma model, isovitexin further induced apoptosis and caused epigenetic regulation through the DNA methyltransferase 1 (DNMT1)/miR-34a/Bcl-2 axis, causing the suppression of stemness and inducing apoptosis in the spheres derived from osteosarcoma cells. Isovitexin also decreased tumor growth and size in a murine xenograft model [401]. In liver cancer, isovitexin decreased sphere and colony formation rates and stemness-associated markers by downregulating FoxM1 via inhibition of MnSOD. Additionally, isovitexin inhibited liver tumor growth in a murine model [406]. In another hepatocarcinoma model, isovitexin caused miR-34a upregulation, which induced apoptosis and suppressed the stemness of SK-Hep-1 spheroids [404].
4.8. Quercetin
Quercetin, a flavonol, is commonly present in red onions, kale, grapes, berries, cherries, broccoli, citrus fruits, as well as in a variety of leaves, seeds, and grains. Quercetin is one of the most abundantly consumed dietary flavonoids, and has a bitter flavor [436]. A recent review by Tang et al. [437] has outlined in detail the anti-cancer actions of quercetin on many types of cancer including, breast, prostate, leukemia, ovarian, gastric, osteosarcoma, melanoma, glioma, lung, colon, and liver in vitro and in vivo. The review highlights the ability of quercetin to inhibit the cell cycle, induce apoptosis through the intrinsic and extrinsic pathways, and inhibit angiogenesis and metastasis in vitro. Quercetin was shown to have the ability to decrease tumor volume and increase survival rate of tumor bearing animals in vivo through apoptosis, and inhibit proliferation, angiogenesis and metastasis [437]. To complement this recent review, we will focus on the anticancer effects of quercetin demonstrated after its publication.
Quercetin treatment reduced cell viability and induced apoptosis in primary and metastatic colon adenocarcinoma cell lines [407]. Quercetin dose-dependently suppressed HGF- and TGFα-induced migration of hepatocellular carcinoma cells by inhibiting the signaling pathway of AKT, but not p38 MAPK [413]. In non-small cell lung carcinoma models, quercetin inhibited proliferation and anchorage-independent growth by inhibiting the Src-mediated Fn14/NF-ĸB pathway both in vitro and in vivo [408] and improved the radiosensitivity of these cells by altering the expression of miR-16-5p and Wee1 [414]. In vivo, lung adenocarcinoma appearance was delayed and increased non-neoplastic body weight gain in mice with tumor oxidative stress after daily treatment with quercetin was observed [420]. In a melanoma model, quercetin treatment decreased proliferation and promoted apoptosis in vivo and in vitro and was found to upregulate IFNα and IFNβ expression through activation of the retinoic acid-inducible gene I promoter [409]. In human prostate cancer cells, in vitro exposure to quercetin downregulated the expression of Metastasis Associated Lung Adenocarcinoma Transcript 1 (MALAT1), suppressed epithelial to mesenchymal transition, promoted apoptosis and inactivated the PI3K/Akt signaling pathway in both a time and dose dependent manner. Similarly, quercetin inhibited the proliferation of prostate cancer in a xenograft model [411,412].
Quercetin in combination with various compounds has been shown to produce enhanced cytotoxic effects in various cancer cells. When combined with the polyphenol resveratrol, quercetin inhibited cell growth and DNA damage, induced S phase cell cycle arrest and caused cell death in oral and pharyngeal cancer cells in a synergistic manner [410]. The combination of curcumin and quercetin lead to synergistic alterations in the expression of genes related to proliferation, apoptosis, cell cycle, inflammation, hypoxia and oxidative stress in myeloid leukemia cancer cells [438] and increased anticancer effects including apoptosis and reactive oxygen species production in breast cancer cells when loaded into apoferritin nanoparticles compared to either free compound alone [439]. Quercetin in combination with Lycopodium clavatum extract resulted in reduced cell growth in colon cancer cells. The combined treatment impacted mRNA expression of pro-apoptotic proteins [440].
Quercetin has been shown to have anticancer effects in cell lines resistant to traditional chemotherapeutic agents and in some cases improved the cytotoxicity of those agents. When combined with paclitaxel in a prostate cancer model, quercetin reduced cell proliferation and migration, while apoptosis, G2/M cell cycle arrest, ER stress and ROS were increased. Beneficial effects in a prostate cancer murine model were also observed, where co-treatment of quercetin with paclitaxel increased the effects of paclitaxel, while causing nearly no additional side effects [416]. The combination of doxorubicin and quercetin also produced favorable effects. In colon adenocarcinoma cells overexpressing the P-gp, quercetin improved the cytotoxicity of doxorubicin by inhibiting the ATP-driven transport activity of P-gp and increasing the intracellular accumulation of doxorubicin. It also downregulated the expression of the glutamine transporter solute carrier family 1, member 5 (SLC1A5) [417]. In multi-drug resistant breast cancer cells, the combination of doxorubicin with quercetin reduced cell viability and was mediated through doxorubicin-induced apoptosis. This was shown to decrease in vivo xenograft tumors without producing toxic effects [418]. Similarly, when combined with docetaxel, quercetin caused a reversal of docetaxel resistance in prostate cancer cells through decreased activation of the androgen receptor and PI3K/Akt pathway, fewer mesenchymal and stem-like cell phenotypes and lower P-gp expression. Features altered after treatment included decreased proliferation, colony formation, migration, invasion, and apoptosis. In vivo, xenograft tumors treated with the combination of quercetin and docetaxel had decreased growth [411]. Treatment with quercetin also decreased lymphoid enhancer-binding factor-1 (Lef1) in docetaxel-resistant breast cancer cells and re-sensitized these cells to docetaxel, acting synergistically to reduce the viability of the drug-resistant cells [415]. Quercetin acted in vitro in HeLa cells and in vivo as an inhibitor of the breast cancer resistance protein (BCRP) [421] while it induced cell death in HL60 cells and multidrug resistant HL60/VINC cells [441]. The cell death was mediated through cell cycle arrest, production of reactive oxygen species, caspase mediated apoptosis, and lysosome membrane permeabilization-dependent mechanisms. Finally, Quercetin exhibited cytotoxic and pro-apoptotic effects on gemcitabine-resistant hepatocellular carcinoma and pancreatic cancer cells. The combined treatment of quercetin and gemcitabine lead to an increased pro-apoptotic response, particularly through S phase cell cycle arrest, the upregulation of tumor protein p53 and downregulation of cyclin D1 [419].
5. Entourage Effect
It was first suggested by Drs. Mechoulam and Ben-Shabat that the endocannabinoid system demonstrated an effect known as the entourage effect, where a multitude of metabolites and related molecules modified the activity of the endogenous cannabinoids anandamide and 2-arachidonoylglycerol [442]. This proposed concept was further extended to explain how whole botanical drugs were often more effective than their isolated components alone [443,444,445,446], but not everyone is convinced of the existence of this effect. The critics describe this effect as a claim with ill-defined and unsubstantiated pharmacological activities that is “used toward the popularization and sale of ostensible therapeutic products” [447]. It is quite possible that both points of view hold some portion of the truth; where one group focuses on the potential of the multiplicity of compounds resulting in synergistic effects from the compounds present in cannabis despite the effects being mechanistically not well characterized, while the other group observes the lack of extensive research but notices over-interpretations of the results and claims that provide high hopes therapeutically. These claims are supported by recent studies that do not show any effect at the cannabinoid receptors by terpenes. In one study, it was shown that none of the five terpenes tested (myrcene, α- and β- pinene, β-caryophyllene, and limonene; either alone or in mixtures) had direct interactions with CB1 or CB2 receptors [448]. Similarly, another study showed that α-pinene, β-pinene, β-caryophyllene, linalool, limonene, and β-myrcene (up to 30–100 μM) could not directly activate CB1 or CB2, or modulate the signaling of the phytocannabinoid agonist Δ9-THC. This suggests that if a phytocannabinoid-terpenoid entourage effect exists, it is not via actions at the CB1 or CB2 receptor level. However, since the reading used involved potassium channels in this study, it remains possible that some of the terpenoids tested could act through other CB1 or CB2-dependent signaling pathways that do not involve potassium channels or simply act through other molecular targets [449]. The hypothesis regarding other targets seems quite plausible; in models of breast cancer, a comparison of pure THC and a botanical extract showed that the botanical drug preparation was more potent than pure THC in producing antitumor responses. The authors reported that the increased potency was not due to the presence of the 5 most abundant terpenes in the preparation (β-caryophyllene, α-humulene, nerolidol, linalool and β-pinene) and that the effects of the botanical preparation modulated different targets and used different mechanisms of action [38].
6. Conclusions
Studies of individual pure compounds found in cannabis have demonstrated, as highlighted in this review article, that many of the compounds present in cannabis could be part of a therapeutic solution for specific problems found during cancer treatment. Cannabis has a number of potential benefits especially in the management of symptoms for patients living with and beyond cancer. Cannabis is useful for various symptoms present during cancer like chemotherapy-induced nausea and vomiting, pain and insomnia, for example. While cannabis may be less potent than some other antiemetics, it is sometimes the only drug that works, and it is the only antiemetic that also increases appetite. The potential of being able to use a single preparation that could hold benefit in the treatment of several adverse effects instead of multiple prescriptions that might interact with each other or with cancer-directed therapies seems advantageous.
It is unlikely that consumption of cannabis is sufficient to act as a stand-alone therapy, but rather, formulations that include compounds present in cannabis could very likely generate the beneficial therapeutic effects needed. There are currently several issues that limit the efficacy of anticancer therapies, such as chemotherapeutic resistance, localization of the tumour in hard to reach tissues, or the severe adverse effects of current therapeutic agents. In each of these cases, cannabis could potentially yield effects that could contribute to the efficacy of a therapy or reduce a side effect profile. Examples of this are found in studies that looked at the effects of several flavonoids and some other compounds that were found to block various effector pumps that are often associated with multidrug resistance. Additionally, some terpenes, like borneol, could facilitate the passage of various formulations past the blood-brain barrier for cancers within the brain tissues.
One of the important considerations when suggesting these compounds as therapeutics lies in their potential effects on the activity of not only known cancer therapeutics, but also other compounds found in cannabis. It has been shown that co-treatment of either THC or CBD with carfilzomib can lower the concentration required to have an effect on the viability and migration of cancer cells [77]. Importantly, when the THC and CBD were used in combination the levels of both compounds required to have an effect on carfilzomib was reduced. This then suggests potential synergistic effects of both the cannabinoids with the therapeutic and with each other. As multiple compounds, whether cannabinoids, terpenes or flavonoids have also been shown to display synergistic effects with current chemotherapeutic agents, this may allow for a reduced dosage of each agent required to produce a therapeutic effect, which has the potential to decrease adverse effects experienced by patients from treatments. As noted by some of the critics of this purported entourage effect, there may also be an exacerbation of negative effects, but this would clearly be evaluated within a systematic evaluation of the costs/benefits of combinatorial therapies. Some studies have begun evaluating the potential correlations of different compounds being produced together within cannabis cultivars [322,450,451,452]. It may be possible that clusters of compounds are required to act synergistically. These combinations have given plants unique profiles that may make some better therapeutics. By analyzing the specific chemical characteristics of the most effective cultivars, it may be possible to define and refine the chemicals that are responsible for their therapeutic benefits. This may provide a basis for the optimization of the ratios of compounds used as polytherapies. This is still highly speculative, and it may simply be that the wrong combination of compounds in botanical preparations were used, lacking flavonoids, for example.
With most of the studies up to now having been done in cell lines or animal models, a lot of work remains, in particular in regard to the bioavailability of these plant-derived compounds, before we fully understand the potential benefits of the cannabis polypharmacy in a way that could be used for the treatment of cancer in humans. Additional clinical studies are needed to clarify whether some of these compounds (alone or in combination with other anticancer agents) could be useful in anticancer therapies.
Author Contributions
Writing—original draft preparation, A.M.T., E.G.W., D.J.D.; writing—review and editing, L.D.E., D.J.D., A.M.T., E.G.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by Beatrice Hunter Cancer Research Institute Seed funding and Dalhousie University Faculty of Medicine Bridge Funding. A.M.T. was supported by NSERC USRA studentship. E.G.W. is supported by a Dalhousie University Department of Pharmacology graduate bursary.